Most of the genomes available in the databases are not complete because extracting complete genomes is tricky and complicated. Repeated sections in the genomes and the sequencing technologies have some drawbacks. Even long-read sequencing techniques are not enough. Therefore, hybrid assembly methods which combine short and long-read technologies are great for obtaining complete genomes.
There was an isolated bacterium in our lab that had novel abilities. Illumina Sequencing was used to obtain its partial genome five years ago (There were more than 100 contigs, so it was fragmented). Getting a higher quality genome of it was crucial, so we wanted to create the complete genome with a hybrid assembly method. Therefore, we sequenced it again with Oxford Nanopore to obtain longer reads. Besides, the bacterium was in the deep freeze all the time, so we expected to see almost the same DNA even though there were some small mutations.
The hybrid assembly method actually gave good results in creating the circular chromosomal DNA; also, there was no plasmid in the bacterium. However, there were some exciting things in the assembly graph (Figure 1). The red-colored segment named C131 was unconnected, and it was standing alone. More interestingly, it wasn’t circular, and it was tiny (2832 bp). A different number of inputs was made, but for all of them, it was still the same. It did not have an insertion-related sequence in it. Also, it did not have a repeating section. The functional analysis showed it has a partial RND efflux gene. Why was the partial RND efflux gene standing alone outside of the chromosome?

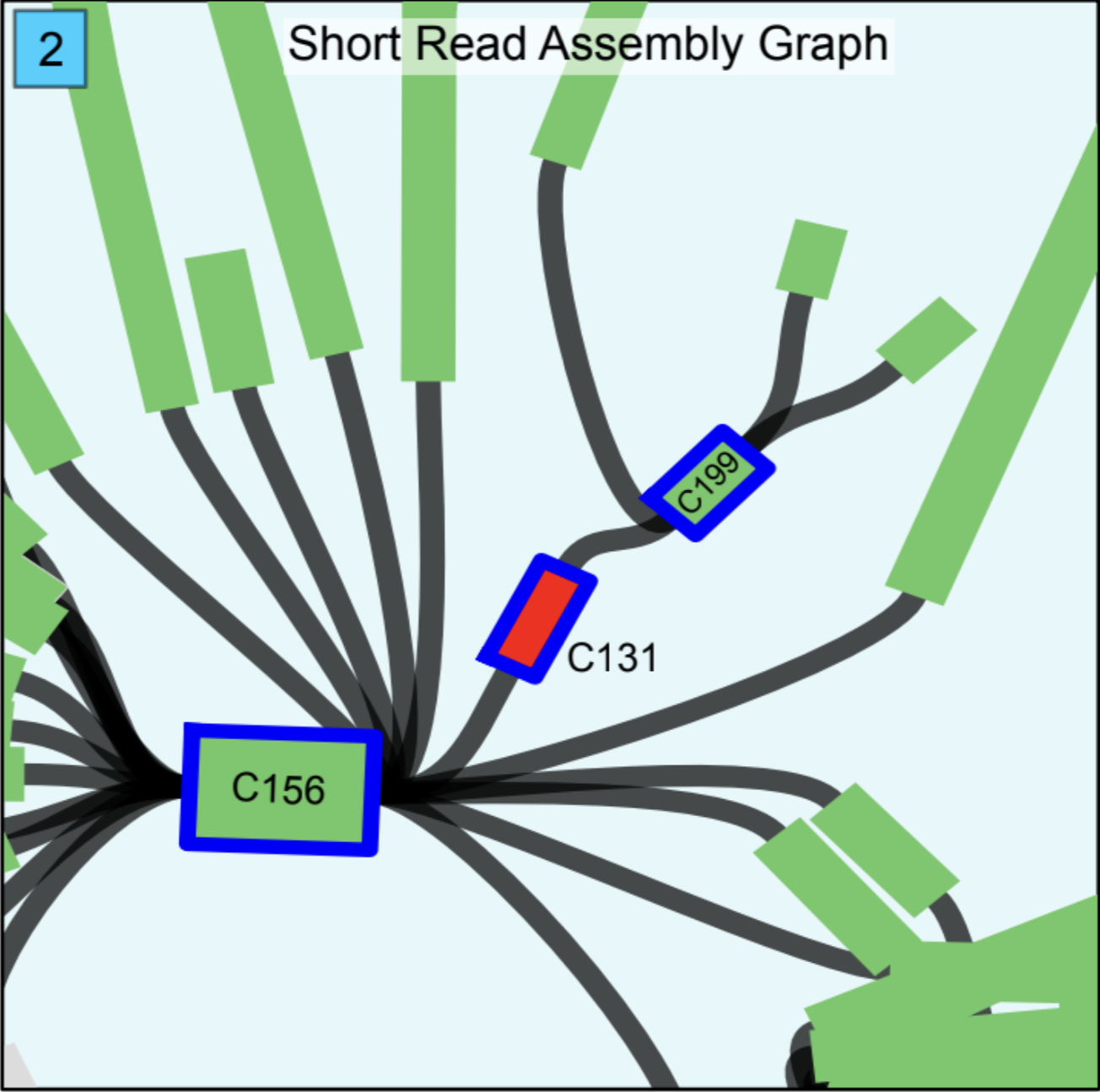
Assembly graphs can be helpful to see what is going on. It is an intermediate step in the assembly process. Also, it has information on how contigs are connected to each other. The assembly graph of just Illumina short reads was investigated. The C131 was in the genome according to the assembly graph because it was connected to the other two contigs named C156 and C199 (Figure 2). Therefore, something changed before Nanopore sequencing.
A BLAST search was performed, the query was C131, and the subject was the circular chromosome of the hybrid assembly. According to the BLAST result, C131 was cut into two pieces in the genome, and there was a gap between the two pieces (Figure 3). According to the functional analysis, the gap is actually an Insertion Sequence (IS). Also, there were duplicated nucleotides in BLAST results because the insertion event created Direct Repeats (DR). DR is also the insertion site of the IS that is “TTAG” (Figure 4).


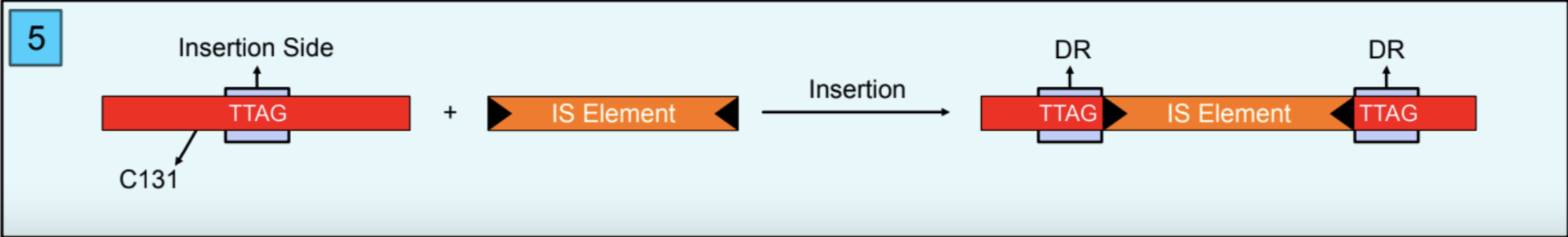
As a final, C131 was in the genome but with an IS in it (Figure 5), so the hybrid method could not insert it. The explanation for this situation is that the genome changed during sequencing with an IS insertion. Bacterial genomes are elastic and there can be transposition events even in short times. The changes in the genome can be massive (more than 1kb) and not like mutations because of DNA replication so that we can see them like artifacts in assembly processes, but actually, they are not.
- Altınbağ, R. C., 2021. Complete Genome Sequencing and Analyzing the Genes of Pseudomonas sp. BIOMIG, M.S. Thesis, Boğaziçi University
- Altinbag, R. C., Ertekin, E., & Tezel, U. (2020). Complete Genome Sequence of Pseudomonas Sp. Strain BIOMIG1BAC, Which Mineralizes Benzalkonium Chloride Disinfectants. Microbiology Resource Announcements, 9(20), e00309-20.